FENYLOKETONURIA – CZY TWOJE DZIECI BĘDĄ ZDROWE?

Fenyloketonuria (ang. phenylketonuria, PKU) to najczęstsza wrodzona choroba dotycząca metabolizmu aminokwasów. W Polsce co 46 osoba nie zdaje sobie sprawy z tego, że jest zdrowym nosicielem nieprawidłowego genu PKU, który może przyczynić się do poważnej choroby potomstwa. Rocznie na terenie Polski rodzi się około 60-70 dzieci dotkniętych tą wadą.
Wcześnie niewykryta i nieleczona fenyloketonuria może prowadzić do poważnych komplikacji zdrowotnych oraz problemów z intelektem i funkcjonowaniem poznawczo-behawioralnym.
Zaburzona przemiana fenyloalaniny
Fenyloketonuria jest chorobą, w której następuje całkowity brak lub znaczne ograniczenie aktywności enzymu hydroksylazy fenyloalaninowej (PAH).
Enzym ten odpowiada za przemianę w komórkach wątroby aminokwasu fenyloalaniny w tyrozynę.

W konsekwencji zaburzeń tego procesu poziom fenyloalaniny we krwi wzrasta, przedostaje się do ośrodkowego układu nerwowego i tam powoduje poważne uszkodzenia.
Z kolei ilość tyrozyny niezbędnej do budowy hormonów tarczycy, nadnerczy, białek ustrojowych oraz wytwarzania melaniny maleje i staje się niewystarczająca dla prawidłowej pracy organizmu.
Uwarunkowania genetyczne
Jak już wcześniej wspomnieliśmy, fenyloketonuria jest chorobą uwarunkowaną genetycznie, przekazywaną dziecku przez rodziców, w której dochodzi do mutacji genów znajdujących się na 12 chromosomie.
Dziedziczona jest autosomalnie recesywnie, co oznacza, że do pojawienia się choroby potrzebny jest jeden nieprawidłowy gen wywołujący fenyloketonurię od ojca i jeden takiż sam gen od matki.

Rodzice dzieci chorych na fenyloketonurię rzadko kiedy zdają sobie sprawę z posiadania nieprawidłowego genu i możliwości przekazania swemu dziecku ciężkiej choroby.
Są bezobjawowymi nosicielami jednego genu „chorego” i jednego genu „zdrowego”, dlatego też choroba ta nie daje u nich żadnych objawów.
Jeżeli matka i ojciec są zdrowymi nosicielami nieprawidłowego genu, istnieje 25% ryzyka, że każde z ich potomstwa urodzi się chore.
Jeśli jeden rodzic choruje na fenyloketonurię, a drugi jest jej bezobjawowym nosicielem, istnieje 50% ryzyka, że każde ich dziecko urodzi się chore.
Jeżeli natomiast i matka, i ojciec chorują na fenyloketonurię pewność, że ich dzieci urodzą się chore wynosi 100%.
Wczesne i późne objawy
Zazwyczaj pierwsze objawy fenyloketonurii pojawiają się około trzeciego miesiąca życia dziecka i zalicza się do nich:
• zaburzenia w rozwoju psychoruchowym;
• charakterystyczny „mysi” zapach potu;
• osłabienie koncentracji;
• zmniejszone lub zwiększone napięcie mięśniowe;
• jasnoniebieskie oczy, jasne włoski i bladą cerę;
• nadpobudliwość;
• niekiedy pojawia się wysypka na skórze, wymioty oraz drgawki.
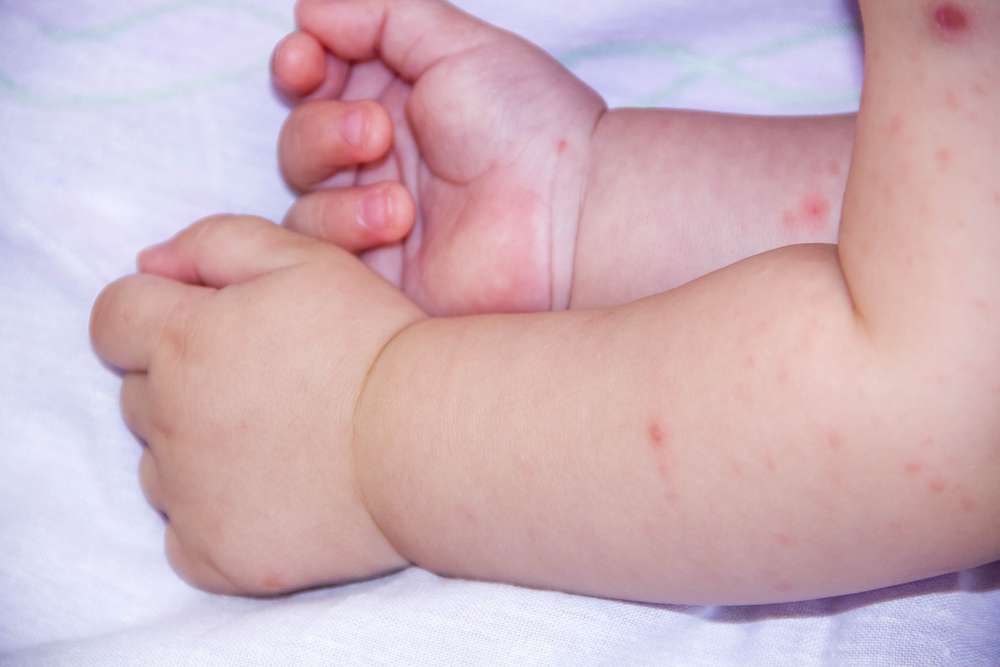
Z czasem objawy mogą się nasilać.
Dużym zagrożeniem dla prawidłowego rozwoju dziecka i późniejszego funkcjonowania w normie intelektualnej jest lekceważenie fenyloketonurii i jej nieleczenie.
Wśród najczęściej występujących objawów nieleczonej lub zbyt późno leczonej choroby znajdują się:
• niepełnosprawność intelektualna – najczęściej upośledzenie umysłowe w stopniu głębokim;
• lekoodporna padaczka;
• postępujące zaburzenie neurologiczne, np. niezborność ruchów lub drżenie kończyn;
• małogłowie;
• wypryski skórne;
• zachowania specyficzne dla dzieci autystycznych, np. lęk przed obcymi osobami;
• niekontrolowane wybuchy złości, nieuzasadnione napady agresji, autodestrukcja;
• zaburzenia mowy;
• problemy z chodzeniem;
• zaburzenia snu;
• problemy z uwagą i koncentracją;
• psychozy.
Fenyloketonuria i dieta
Sekretem leczenia fenyloketonurii jest odpowiednia dieta tzw. dieta niskofenyloalaninowa i utrzymywanie odpowiedniego stężenia fenyloalaniny we krwi.
Głównym źródłem fenyloalaniny są białka pokarmowe zawarte w wysokobiałkowych produktach np. mięsie, rybach, mleku i przetworach mlecznych, jajkach, przetworach zbożowych, kakao, orzechach, czekoladzie, soi, fasoli i grochu.
Należy przestrzegać diety eliminacyjnej, która pozwoli utrzymać minimalny, ale konieczny do prawidłowego funkcjonowania całego organizmu poziomu fenyloalaniny u chorego na PKU.
Maksymalny poziom fenyloalaniny u dziecka zdrowego wynosi 2 mg/100 ml krwi.
U dziecka chorego stężenie waha się w zależności od jego wieku.
Dla przedziału od 0 do 5 lat wynosi 2-4 mg%; od 6 do 14 lat wynosi 2-8 mg%, a powyżej 14 lat wynosi mniej niż 12 mg%.
Przeprowadzanie badań kontrolnych w celu oznaczania stężenie fenyloalaniny we krwi należy przeprowadzać z następującą częstością:
• od 0 do 12 miesiąca życia dziecka – raz w tygodniu;
• do 3 roku życia dziecka – co tydzień lub dwa;
• od 3 do 7 lat – co dwa tygodnie;
• powyżej 7 lat – raz w miesiącu.
Tylko rygorystyczne przestrzeganie diety daje możliwość prawidłowego rozwoju na gruncie fizycznym, emocjonalnym i intelektualnym.
Wykrywanie fenyloketonurii
Od 1965 roku w Polsce trwa program, w którym wszystkie noworodki podlegają przeprowadzeniu testów przesiewowych.
Testy wykrywające fenyloketonurię przeprowadza się w czwartej dobie życia dziecka.

Badanie jest małoinwazyjne, polega jedynie na pobraniu odrobiny krwi na specjalną bibułkę i oznaczeniu poziomu stężenia fenyloalaniny.
Ważna jest jednak przede wszystkim profilaktyka przed poczęciem.
Przyszłym rodzicom zaleca się wykonywanie badań genetycznych, które umożliwiają wykrycie fenyloketonurii i wdrożenie odpowiednich działań profilaktycznych.
Nieleczona i niekontrolowana fenyloketonuria u ciężarnej kobiety stwarza duże ryzyko wad serca u płodu, upośledzeń umysłowych oraz mikrocefalii.
Dlatego też konieczne jest w tym przypadku świadome, wcześniejsze przygotowywanie organizmu do zajścia w ciążę.
*Niebieską czcionką zaznaczono odnośniki np. do badań, tekstów źródłowych lub artykułów powiązanych tematycznie.
P.S. Informacje przedstawione w artykule nie są pisane przez lekarza. Nie są one fachową opinią, ani poradą medyczną. Nie mogą zastąpić opinii i wiedzy pracownika służby zdrowia, np. lekarza. Wszelkie rady, które są na mojej stronie stosujesz wyłącznie na własną odpowiedzialność.
https://www.youtube.com/watch?v=IViY-A0D-w4
https://www.youtube.com/watch?v=nRplapxcjg0
https://www.youtube.com/watch?v=Qsjbx4aPbI8


